VASP Installations on Tetralith & Sigma
First of all, VASP is a licensed software, your name needs to be included on a VASP license in order to use NSC’s centrally installed VASP binaries. Read more about how we handle licensing of VASP at NSC.
Some problems which can be encountered running VASP are described at the end of this page.
VASP6
VASP6 was released in beginning of 2020. This means e.g. that VASP5 license holders will need to update their license in order to access VASP6 installations at NSC. If you have a VASP 5.4.4 license, you are typically covered for updates of VASP 6.X.X for three years, check your license for the exact details.
Documentation
For using VASP, refer to the extensive VASP wiki which includes many examples and topics on how to run VASP. It can also be very useful to check the forum and other resources available at the VASP webpage.
The new features of VASP6 are described in the VASP wiki.
From time to time NSC will arrange seminars or brief workshops on VASP, with focus typically on how to get started using it on our supercomputers. See the events or past events pages, here is a link to the latest event which contains e.g. the presentations.
How to run: quick start
A minimum batch script for running VASP looks like this:
#!/bin/bash
#SBATCH -J jobname
#SBATCH -N 4
#SBATCH --ntasks-per-node=32
#SBATCH -t 4:00:00
#SBATCH -A SNIC-xxx-yyy
module add VASP/5.4.4.16052018-nsc1-intel-2018a-eb
mpprun vasp_[std/gam/ncl]
This script allocates 4 compute nodes with 32 cores each, for a total of 128 cores (or MPI ranks) and runs VASP in parallel using MPI. Note that you should edit the jobname and the account number before submitting.
For best performance, use these settings in the INCAR file for regular DFT calculations:
NCORE=32 (or the same number as you use for --ntasks-per-node)
NSIM=16 (or higher)
For hybrid-DFT calculations, use:
NCORE=1
NSIM=16 (the value is not influential when using e.g. ALGO=damped)
POTCAR
The latest version of the PAW potential files, POTCARs, can be found here:
/software/sse2/tetralith_el9/manual/vasp/POTCARs
Previous versions are available here:
/software/sse/manual/vasp/POTCARs
For changes and updates, refer to the README.UPDATES file in the respective folders. Read more on which PAW potentials are recommended.
Modules
NSC’s VASP binaries have hard-linked paths to their run-time libraries, so you do not need to load a VASP module or set LD_LIBRARY_PATH and such for it work. We mainly provide modules for convenience, as you do not need to remember the full paths to the binaries. Though if you directly run the binary for non-vanilla installations with intel-2018a, you need to set in the job script (see details in section “Problems” below):
export I_MPI_ADJUST_REDUCE=3
This is not needed for other modules built with e.g. intel-2018b.
There is generally only one module of VASP per released version directly visible in the module system. It is typically the recommended standard installation of VASP without source code modifications. Load the VASP module corresponding to the version you want to use.
module add VASP/5.4.4.16052018-nsc1-intel-2018a-eb
Then launch the desired VASP binary with “mpprun”:
mpprun vasp_std
Special builds
If you have a license and need a special build of VASP, you are welcome to contact support. Special builds do not always have their own module (remember to set export I_MPI_ADJUST_REDUCE=3). You can find some of these builds if you go to the VASP version installation folder on Tetralith/Sigma. For example, for VASP 6.2.1 check under
/software/sse/manual/vasp/6.2.1.29042021/intel-2018a-omp
here you can find the folders
nb96/nsc1
w90/2.1/nsc1
w90/3.1/nsc1
which are special builds for (1) setting NB=96 in scala.F, useful for large jobs, (2) for use together with Wannier90 2.1 Wannier90/2.1.0-nsc1-intel-2018a-eb, (3) for use together with Wannier90 3.1, Wannier90/3.1.0-nsc1-intel-2018a-eb.
Naming scheme and available installations
The VASP installations are usually found under /software/sse/manual/vasp/version/buildchain/nsc[1,2,...]/. Each nscX directory corresponds to a separate installation. Typically, the higher build with the latest buildchain is the preferred version. They may differ by the compiler flags used, the level of optimization applied, and the number of third-party patches and bug-fixes added on.
Each installation contains three different VASP binaries:
- vasp_std: “normal” version for bulk system
- vasp_gam: gamma-point only (big supercells or clusters)
- vasp_ncl: for spin-orbit/non-collinear calculations
We recommended using either vasp_std or vasp_gam if possible, in order to decrease the memory usage and increase the numerical stability. Mathematically, the half and gamma versions should give identical results to the full versions, for applicable systems. Sometimes, you can see a disagreement due to the way floating point numerics works in a computer. In such cases, we would be more inclined to believe the gamma/half results, since they preserve symmetries to a higher degree.
Constrained structure relaxation
Binaries for constrained structure relaxation are available for some of the modules. The naming scheme is as follows, e.g.:
- vasp_x_std: relaxation only in x-direction, yz-plane is fixed
- vasp_xy_std: relaxation only in xy-plane, z-direction is fixed
Note: This naming scheme gives the relaxation direction.
vdW-kernel
The VASP binaries contain a hard-coded path to the vdW_kernel.bindat file located in the file system under /software/sse/manual/vasp, so that you do not need to generate it from scratch. From 6.4.3 it’s instead generated efficiently when running VASP.
OpenMP threading
Several VASP6 installations are compiled with support for optional OpenMP threading. It might e.g. be useful for increasing the efficiency when reducing the number of tasks (mpi ranks) per node in order to save memory.
To use the OpenMP threading, compare with the example job script from the top of the page, now setting 4 OpenMP threads. Note the reduced tasks per node (mpi ranks) such that 8 tasks x 4 OpenMP threads = 32 (cores/node). Also note the setting of OMP_NUM_THREADS and OMP_STACKSIZE
#!/bin/bash
#SBATCH -J jobname
#SBATCH -N 4
#SBATCH --ntasks-per-node=8
#SBATCH -t 4:00:00
#SBATCH -A SNIC-xxx-yyy
export OMP_NUM_THREADS=4
export OMP_STACKSIZE=512m
module add VASP/6.3.1.04052022-omp-nsc1-intel-2018a-eb
mpprun vasp_[std/gam/ncl]
Note: for the special case of two OpenMP threads, OMP_NUM_THREADS=2, and running on more than one node, use the following command instead of mpprun:
srun --mpi=pmi2 -m block:block vasp_[std/gam/ncl]
VASP 6.4.3
The update 6.4.3 includes new features (e.g. interface to libMBD), several improvements and bug fixes, see link for all details.
omp-hpc1-intel-2023a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory.
module add VASP/6.4.3.19032024-omp-hpc1-intel-2023a-eb
mpprun vasp_std
For this new build (previous are from old CentOS 7), it wasn’t needed to set I_MPI_ADJUST_REDUCE to avoid BRMIX problems.
VASP 6.4.2
The update 6.4.2 includes several fixes, mostly related to the ML part, see link for details. Note, for ML_ISTART=2 support at the moment use the intel-2021.3.0-oneapi build!
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory. Note this build has problem with ML_ISTART=2.
module add VASP/6.4.2.20072023-omp-nsc1-intel-2018a-eb
mpprun vasp_std
For ML_ISTART=2 to work, at the moment use the intel-2021.3.0-oneapi build (with HDF5 support and OpenMP switched off), e.g.:
mpprun /software/sse/manual/vasp/6.4.2.20072023/intel-2021.3.0-oneapi/nsc1/vasp_std
VASP 6.4.1
The update 6.4.1 includes several fixes and improvements, most related to the ML part, see link for details. Note, for ML_ISTART=2 support at the moment use the intel-2021.3.0-oneapi build!
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory. Note this build has problem with ML_ISTART=2.
module add VASP/6.4.1.07042023-omp-nsc1-intel-2018a-eb
mpprun vasp_std
For ML_ISTART=2 to work, at the moment use the intel-2021.3.0-oneapi build (with HDF5 support and OpenMP switched off), e.g.:
mpprun /software/sse/manual/vasp/6.4.1.07042023/intel-2021.3.0-oneapi/nsc2/vasp_std
Here nsc2 includes additional fixes compared with nsc1 for ML calculations (for exact details, refer to “build.txt” in respective folders).
VASP 6.4.0
The update 6.4.0 includes several changes and additions for DFT and hybrid functionals, ACFDT and GW, ML force fields have new fast-prediction mode with 20-100 times faster MD trajectories possible, see link for details. Note, for ML_ISTART=2 support at the moment use the gcc-2022a build!
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory. Note this build has problem with ML_ISTART=2.
module add VASP/6.4.0.14022023-omp-nsc1-intel-2018a-eb
mpprun vasp_std
For ML_ISTART=2 to work, at the moment use the gcc-2022a build, e.g.:
mpprun /software/sse/manual/vasp/6.4.0.14022023/gcc-2022a-omp/nsc1/vasp_std
VASP 6.3.2
The update 6.3.2 includes e.g. a bug fix related to restart of ML training runs, calculating total number of GPUs and assorted minor bugfixes and improvements. All tests in the included testsuite goes through.
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory.
module add VASP/6.3.2.27062022-omp-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.3.1
The update 6.3.1 includes a number of fixes, e.g. for bandstructure calculations with hybrid functionals, machine learning + parallel tempering, tetrahedon method + k-point grids, bug which caused RPA force calculations to sometimes crash, and improved stability of machine learning algorithm by new default settings. All tests in the included testsuite goes through.
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory.
module add VASP/6.3.1.04052022-omp-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.3.0
The update 6.3.0 includes machine learning force fields (of great interest for MD), single-shot computation of bandstructures (KPOINTS_OPT), improved support for libxc XC-functional library, input and output from HDF5 files, and a number of other additions, performance optimizations and bug fixes. All tests in the included testsuite goes through.
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory.
module add VASP/6.3.0.20012022-omp-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.3.0 utility installation with VTST and BEEF
VASP built in the same way as the regular version, but including VTST 4.2 (vtstcode-184, vtstscripts-978 and BEEF. For more information, check the respective links.
module add VASP-VTST/4.2-6.3.0.20012022-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.2.1
The update 6.2.1 includes some bugfixes; an error in the RSCAN functional, XAS implementation, crash for vasp_gam using Wannier90 interface. An improvement is that NCCL again is made optional for the OpenACC version. All tests in the included testsuite goes through.
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading. In brief, OpenMP might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory.
module add VASP/6.2.1.29042021-omp-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.2.0
The update 6.2.0 includes, for example, assorted fixes for non-critical issues, compressed Matsubara grids for finite temperature GW/RPA calculations, a cubic-scaling OEP-Method, support for Wannier90 2.X and 3.X, semi-empirical DFTD4 vdW corrections, self-consistent potential correction for charged periodic systems (by Peter Deak et al.). All tests in the included testsuite goes through.
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading.
module add VASP/6.2.0.14012021-omp-nsc1-intel-2018a-eb
mpprun vasp_std
For an example how to use the OpenMP threading, see above.
VASP 6.1.2
The update 6.1.2 fixes a broken constrained magnetic moment approach in VASP6 (ok in VASP5). Also included (assumed from update 6.1.1) is fix for a bug which affected the non-local part of the forces in noncollinear magnetic calculations with NCORE not set to 1 (ok in VASP5).
nsc1-intel-2018a: Compiled in the same way as for 6.1.0 (see below).
module add VASP/6.1.2.25082020-nsc1-intel-2018a-eb
mpprun vasp_std
omp-nsc1-intel-2018a: Compiled with support for optional OpenMP threading, otherwise similar to the above installation. In brief, it might e.g. be useful for increasing the efficiency when you have reduced the number of tasks (mpi ranks) per node in order to save memory. For an example of a job script, see above.
module add VASP/6.1.2.25082020-omp-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 6.1.0
When testing VASP6, I needed to set export I_MPI_ADJUST_REDUCE=3 for stable calculations (done automatically at NSC by loading the corresponding VASP modules). Otherwise, calculations might slightly differ or behave differently (ca. 1 out of 10).
nsc1-intel-2018a: The first VASP6 module at NSC. A build with no modifications of the source code and using more conservative optimization options (-O2 -xCORE-AVX2). This build does not include OpenMP. All tests in the new officially provided testsuite passed.
module add VASP/6.1.0.28012020-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 5.4.4 Patch 16052018
nsc1-intel-2018a: After fixing in the module by automatically setting I_MPI_ADJUST_REDUCE=3 (see discussion in Modules) this is now again the recommended module. VASP built from the original source with minimal modifications using conservative optimization options (-O2 -xCORE-AVX512). The VASP binaries enforce conditional numerical reproducibility at the AVX-512 bit level in Intel’s MKL library, which we believe improves numerical stability with no cost to performance.
module add VASP/5.4.4.16052018-nsc1-intel-2018a-eb
mpprun vasp_std
nsc1-intel-2018b: Built with slightly newer intel compiler. However, it seems more sensitive for certain calculations, leading to crashes, which are avoided by using nsc1-intel-2018a.
module add VASP/5.4.4.16052018-nsc1-intel-2018b-eb
mpprun vasp_std
nsc2-intel-2018a: Due to spurious problems when using vasp_gam it’s compiled differently as compared to the nsc1-intel-2018a build (-O2 -xCORE-AVX2).
module add VASP/5.4.4.16052018-nsc2-intel-2018a-eb
mpprun vasp_std
VASP “vanilla” 5.4.4 Patch 16052018
nsc1-intel-2018a: A special debug installation is available. VASP built with debugging information and lower optimizmation. Mainly intended for troubleshooting and running with a debugger. Do not use for regular calculations, e.g.:
module add VASP/5.4.4.16052018-vanilla-nsc1-intel-2018a-eb
mpprun vasp_std
If you see lots of error messages BRMIX: very serious problems this might provide a solution. OBS: This module doesn’t actually include the latest patch 16052018.
VASP 5.4.4 Patch 16052018 with Wannier90 2.1.0
nsc2-intel-2018a: VASP built for use together with Wannier90 2.1.0. Compared with the previous nsc1 version, it includes a patch for use with Wannier90 by Chengcheng Xiao, see this link for more information. Load and launch e.g. with:
module add VASP/5.4.4.16052018-wannier90-nsc2-intel-2018a-eb
mpprun vasp_std
nsc1-intel-2018a: VASP built for use together with Wannier90 2.1.0. Load and launch e.g. with:
module add VASP/5.4.4.16052018-wannier90-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 5.4.4 Patch 16052018 newer utility installation with VTST and VASPsol++
**hpc1-intel-2023a **: A newer VASP5 build including VTST 3.2 as well as VASPsol++. For more information, check the respective links.
module add VASP-VTST/3.2-sol++-5.4.4.16052018-hpc1-intel-2023a-eb
mpprun vasp_std
VASP 5.4.4 Patch 16052018 utility installation with VTST, VASPsol and BEEF
nsc2-intel-2018a: VASP built in the same way as the regular version, but including VTST 3.2 (vtstcode-176, vtstscripts-935), VASPsol and BEEF. For more information, check the respective links.
module add VASP-VTST/3.2-sol-5.4.4.16052018-nsc2-intel-2018a-eb
mpprun vasp_std
VASP “vanilla” 5.4.4 Patch 16052018 utility installation with VTST, VASPsol and BEEF
nsc1-intel-2018a: A special debug installation is available.
module add VASP-VTST/3.2-sol-5.4.4.16052018-vanilla-nsc1-intel-2018a-eb
mpprun vasp_std
VASP 5.4.4 Patch 16052018 with occupation matrix control modification
nsc1-intel-2018a: Similar to the regular version vasp module nsc2-intel-2018a, but modified for occupation matrix control of d and f electrons. See this link and this other link for more information.
module add VASP-OMC/5.4.4.16052018-nsc1-intel-2018a-eb
mpprun vasp_std
Performance and scaling
In general, you can expect about 2-3x faster VASP speed per compute node vs Triolith, provided that your calculation can scale up to using more cores. In many cases, they cannot, so we recommend that if you ran on X nodes on Triolith (#SBATCH -N X), use X/2 nodes on Tetralith, but change to NCORE=32. The new processors are about 1.0-1.5x faster on a per core basis, so you will still enjoy some speed-up even when using the same number of cores.
Selecting the right number of compute nodes
Initial benchmarking on Tetralith showed that the parallel scaling of VASP on Tetralith is equal to, or better, than Triolith. This means that while you can run calculations close to the parallel scaling limit of VASP (1 electronic band per CPU core) it is not recommended from an efficiency point of view. You can easily end up wasting 2-3 times more core hours than you need to. A rule of thumb is that 6-12 bands/core gives you 90% efficiency, whereas scaling all the way out to 2 bands/core will give you 50% efficiency. 1 band/core typically results in < 50% efficiency, so we recommend against it. If you use k-point parallelization, which we also recommend, you can potentially multiply the number of nodes by up the number of k-points (check NKPT in OUTCAR and set KPAR in INCAR). A good guess for how many compute nodes to allocate is therefore:
number of nodes = KPAR * NBANDS / [200-400]
Example: suppose we want to do regular DFT molecular dynamics on a 250-atom metal cell with 16 valence electrons per atom. There will be at least 2000 + 250/2 = 2150 bands in VASP. Thus, this calculation can be run with up to (2150/32) = ca 67 Tetralith compute nodes, but it will be very inefficient. Instead, a suitable choice might be ca 10 bands per cores, or 2150/10 = 215 cores, which corresponds to around 6-7 compute nodes. To avoid prime numbers (7), we would likely run three test jobs with 6,8 and 12 Tetralith compute nodes to check the parallel scaling.
A more in depth explanation with example can be found in the blog post “Selecting the right number of cores for a VASP calculation”.
What to expect in terms of performance
To show the capability of Tetralith, and provide some guidance in what kind of jobs that can be run and how long they would take, we have re-run the test battery used for profiling the Cray XC-40 “Beskow” machine in Stockholm (more info here). It consists of doped GaAs supercells of varying sizes with the number of k-points adjusted correspondingly.
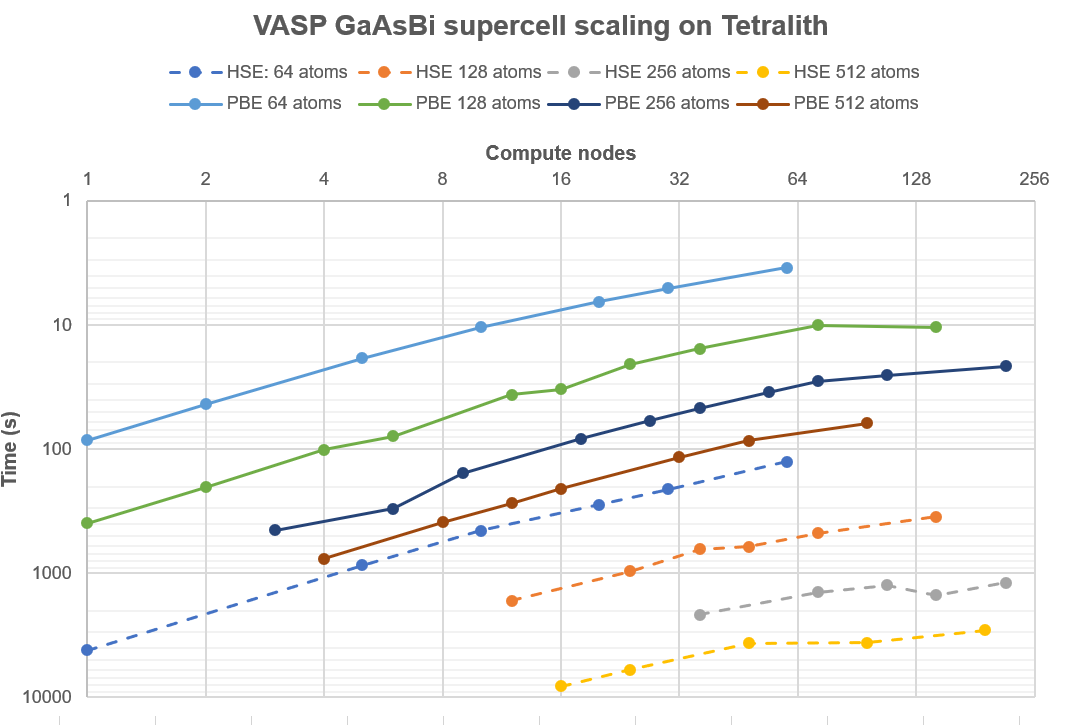
Fig. 1: Parallel scaling on Tetralith of GaAs supercells with 64 atoms / 192 bands, 128 atoms / 384 bands, 256 atoms / 768 bands, and 512 atoms / 1536 bands. All calculations used k-point parallelization to the maximum extent possible, typically KPAR=NKPT. The measured time is the time taken to complete a complete SCF cycle.
The tests show that small DFT jobs (< 100 atoms) run very fast using k-point parallelization, even with a modest number of compute nodes. The time for one SCP cycle can often be less than 1 minute, or 60 geometry optimization steps per hour. In contrast, hybrid-DFT calculations (HSE06) takes ca 50x longer time to finish, regardless of how many nodes are thrown at the problem. They scale somewhat better, so typically you can use twice the number of nodes at the same efficiency, but it is not enough to make up the difference in run-time. This is something that you must budget for when planning the calculation.
As a practical example, let us calculate how many core hours that would be required to run 10,000 full SCF cycles (say 100 geometry optimizations, or a few molecular dynamics simulations). The number of nodes has been chosen so that the parallel efficiency is > 90%:
| Atoms | Bands | Nodes | Core hours |
|---|---|---|---|
| 64 | 192 | 5 | 8,000 |
| 128 | 384 | 12 | 39,000 |
| 256 | 768 | 18 | 130,000 |
| 512 | 1536 | 16 | 300,000 |
The same table for 10,000 SCF cycles of HSE06 calculations looks like:
| Atoms | Bands | Nodes | Core hours |
|---|---|---|---|
| 64 | 192 | 10 | 400,000 |
| 128 | 384 | 36 | 2,000,000 |
| 256 | 768 | 36 | 6,900,000 |
| 512 | 1536 | 24 | 13,000,000 |
For comparison, a typical large SNAC project might have an allocation of 100,000-1,000,000 core hours per month with several project members, while a smaller personal allocation might be 5,000-10,000 core hours/month. Thus, while it is technically possible to run very large VASP calculations quickly on Tetralith, careful planning of core hour usage is necessary, or you will exhaust your project allocation.
Another important difference vs Triolith is the improved memory capacity. Tetralith has 96 GB RAM memory node (or about 3 GB/core vs 2 GB/core on Triolith). This allows you to run larger calculations using less compute nodes, which is typically more efficient. In the example above, the 512-atom GaAsBi supercell with HSE06 was not really possible to run efficiently on Triolith due to limited memory.
Compiling VASP from source
Finally, some notes and observations on compiling VASP on Tetralith. You can find makefiles in the VASP installation directories under /software/sse/manual/vasp/.
- Intel’s Parallel Studio 2018 Update 1 compilers and MKL appears to work well. Use the
buildenv-intel/2018a-ebmodule on Tetralith to compile. You can use it even if you compile by hand and not use EasyBuild. - VASP’s default makefile.include can be used, but we strongly recommend to compile with at least
-xCORE-AVX2or-xCORE-AVX512optimization flags for better performance on modern hardware. - Do not compile with
-O3using Intel’s 2018 compiler, stay with-O2. When we tested, it is not faster, but it produces binaries which have random, but repeatable, convergence problems (1 out of 100 calculations or so). - VASP still cannot be compiled in parallel, but you can always try running
make -j4 stdor similar repeatedly. - VASP compiled with gcc, OpenBLAS and OpenMPI runs slower on Tetralith in our tests.
- Compiling with
-xCORE-AVX512and letting Intel’s MKL library use AVX-512 instructions (which it does by default) seems to help in most cases, ca 5-10% better performance. We have seen cases where VASP runs faster with AVX2 only, so it is worth trying. It might be dependent on a combination of Intel Turbo Boost frequencies and NSIM, but remains to be investigated. If you want to try, you can compile with-xCORE-AVX2, and then set the environment variableMKL_ENABLE_INSTRUCTIONS=AVX2to force AVX2 only. This should make the CPU cores clock a little bit higher at the expense of less FLOPS/cycle. - Previosuly, we have turned on conditional bitwise reproducibility at the AVX512 level in the centrally installed binaries on Tetralith. If you compile yourself, you can also enable this at run-time using the
MKL_CBWR=AVX512environment variable. This was more of an old habit, as we haven’t seen any explicit problems with reproducibility so far, but it does not hurt performance. The fluctuations are typically in the 15th decimal or so. Please note thatMKL_CBWR=AVXor similar, severely impacts performance (-20%). - Our local VASP test suite passes for version 5.4.4 Patch #1 with the same deviations as previously seen on Triolith.
Problems
BRMIX: very serious problems the old and the new charge density differ
If you encountered the problem BRMIX: very serious problems the old and the new charge density differ written in the slurm output, with VASP calculations on Tetralith / Sigma for cases which typically work on other clusters and worked on Triolith / Gamma, it might be related to bug/s which was traced back to MPI_REDUCE calls. These problems were transient, meaning that out of several identical jobs, some go through, while others fail. Our VASP modules are now updated to use another algorithm by setting I_MPI_ADJUST_REDUCE=3, which shouldn’t affect the performance. If you don’t load modules, but run binaries directly, set in the job script:
export I_MPI_ADJUST_REDUCE=3
More details for the interested: the problem was further traced down to our setting of the NB blocking factor for distribution of matrices to NB=32 in scala.F. The VASP default of NB=16 seems to work fine, while NB=96 also worked fine on Triolith. By switching off ScaLAPACK in INCAR, LSCALAPACK = .FALSE. it also works. Furthermore, the problem didn’t appear for gcc + OpenBLAS + OpenMPI builds.
Newer VASP modules built with e.g. intel-2018b use VASP default NB=16, while the non-vanilla modules built with intel-2018a has NB=32 set.
internal error in SETUP_DEG_CLUSTERS: NB_TOT exceeds NMAX_DEG
If you find this problem internal error in SETUP_DEG_CLUSTERS: NB_TOT exceeds NMAX_DEG typically encountered for phonon calculations, you can try the specially compiled versions with higher values of NMAX_DEG, e.g.:
mpprun /software/sse/manual/vasp/5.4.4.16052018/intel-2018b_NMAX_DEG/nsc1_128/vasp_std
mpprun /software/sse/manual/vasp/5.4.4.16052018/intel-2018b_NMAX_DEG/nsc1_256/vasp_std
 User Area
User Area

