WIEN2K Installations on Tetralith & Sigma
“The program package WIEN2k allows to perform electronic structure calculations of solids using density functional theory (DFT). It is based on the full-potential (linearized) augmented plane-wave ((L)APW) + local orbitals (lo) method, one among the most accurate schemes for band structure calculations. WIEN2k is an all-electron scheme including relativistic effects and has many features.”
To use the NSC installations one needs to be covered by a valid license, for more information see this link.
Note: At the moment it doesn't work to run over several nodes.
Initial setup
There are some settings which are needed by WIEN2k. This can be done by running userconfig after loading the WIEN2k module, which will put the settings into .bashrc. These settings could also be put into a file which you source before using WIEN2k (source filname). For most purposes, you can just provide the minimal settings:
export SCRATCH=./
export EDITOR="vi"
where the editor also can be set to e.g. “emacs”. Without these settings, e.g. initalizing your calculation with init_lapw might crash.
Example batch script
A batch script for running WIEN2k may look like below, here running MPI over a single node (32 cores). Also note that a few cores might be enough, depending on the job. See examples further below on how the jobscript can be tailored and also consider the benchmarks at the end of this page. The examples show how to set up the creation of appropriate .machines files used by WIEN2k for parallel calculations.
#!/bin/bash
#SBATCH -A naiss-xxx-yyy
#SBATCH -J jobname
#SBATCH -N 1
#SBATCH --exclusive
#SBATCH -t 12:00:00
module add WIEN2K/23.2-hpc1-intel-2023.1.0-oneapi
# export SCRATCH=$SNIC_TMP
# set .machines for parallel job
# lapw0 running on one node
echo -n "lapw0: " > .machines
echo -n $(hostlist -e $SLURM_JOB_NODELIST | tail -1) >> .machines
echo "$i:8" >> .machines
# run one mpi job on each node (splitting k-mesh over nodes)
for i in $(hostlist -e $SLURM_JOB_NODELIST)
do
echo "1:$i:32 " >> .machines
done
echo granularity:1 >> .machines
echo extrafine:1 >> .machines
run_lapw -p
# if you need the vector files for continuing calculations, e.g.
# for DOS, spectrum etc. copy them to your current directory:
# cp $SNIC_TMP/* .
(Note that you should edit the jobname and the account number before submitting.)
Available modules
WIEN2K/23.2-hpc1-intel-2023.1.0-oneapi
Installation of 23.2 release 9/3/2023, which includes fixes and new features. Note that init_lapw works different, batch mode is now default. For the step-by-step initialization, instead use init_lapw -m. This build includes OpenMP threading and ELPA 2022.11.001.
WIEN2K/19.1-hpc1-intel-2023a-eb
Installation of 19.1 release 25/6/2019, including some extra patches. This version includes OpenMP threading (instructions in preparation) and ELPA 2022.11.001.
Retired modules used on CentOS 7
WIEN2k/23.2-nsc1-intel-2018b-eb
Installation of 23.2 release 9/3/2023, which includes fixes and new features. Note that init_lapw works different, batch mode is now default. For the step-by-step initialization, instead use init_lapw -m. This build includes OpenMP threading and ELPA 2019.05.001.
WIEN2k/21.1-nsc1-intel-2018b-eb
Installation of 21.1 release 14/4/2021, which includes patches and e.g. an improvement of the OpenMP parallelization. This build includes OpenMP threading and ELPA 2019.05.001.
WIEN2k/19.1-nsc1-intel-2018b-eb
Installation of 19.1 release 25/6/2019, including some extra patches. This version includes OpenMP threading (instructions in preparation) and ELPA 2019.05.001.
WIEN2k/18.2-nsc2-intel-2018a-eb
A small update of nsc1 version with fix for running over several nodes.
WIEN2k/18.2-nsc1-intel-2018a-eb
Installation of 18.2 release 17/7/2018. This version includes ELPA 2015.11.001.
Further information
-
See the userguide for test examples and much useful information.
-
If setting
export SCRATCH=$SNIC_TMPnote that it might be problematic when using more than one node (if needed, contact NSC support). -
If the vector files are needed for further processing, you need to retrieve them from the temporary directory as the job finishes.
-
For very heavy jobs with several 100s of inequivalent atoms, it might be necessary to specify in the job script:
echo "lapw2_vector_split 4" >> .machines #typical values 2, 4, 6, 8 -
In general, if you have several k-points it makes sense to use the k-point parallelization in WIEN2k. The most efficient way is to calculate 1 k-point per computer core (if memory use is not too big).
-
For many jobs it can be most efficient to run on less than a full node, e.g. allocating 4, 8 or 16 cores on Tetralith/Sigma, see benchmarks below.
-
For tiny jobs (e.g. small unit cells) it is recommended to just do a
serialrun:... #SBATCH -n 1 ... run_lapw -
The use of the ELPA library can be switched to ScaLAPACK, by changing the 2nd line in the file .in1 or .in1c, from
ELPAtoSCALA:7.00 10 4 ELPA pxq BL 64 (R-MT*K-MAX,MAX L IN WF,V-NMT,LIB) --> 7.00 10 4 SCALA pxq BL 64 (R-MT*K-MAX,MAX L IN WF,V-NMT,LIB)
k-point parallelization
k-point parallelization works over one or several nodes, e.g. using 32 cores/node on Tetralith. It is probably the most efficient way to run WIEN2k if you have many k-points. For example:
...
#SBATCH -n 32
...
for i in $(hostlist -e $SLURM_JOB_NODELIST)
do
for j in {1..32}
do
echo "1:$i:1 " >> .machines
done
done
echo granularity:1 >> .machines
echo extrafine:1 >> .machines
...
At the moment it doesn’t work to run the full k-point parallelization over several nodes.
k-point parallelization and MPI
It is possible to combine k-point parallelization with mpi. For instance, if you have 2 k-points instead of 1, it might be more efficient to split the calculations into two:
...
#SBATCH -n 32
...
for i in $(hostlist -e $SLURM_JOB_NODELIST)
do
echo "1:$i:16 " >> .machines
echo "1:$i:16 " >> .machines
done
echo granularity:1 >> .machines
echo extrafine:1 >> .machines
A different example, if there are more k-points. Here the list of k-points is split over two Tetralith nodes (32x2 cores) and then split in 8 parts running on 4 cores at each node:
...
#SBATCH -n 64
...
for i in $(hostlist -e $SLURM_JOB_NODELIST)
do
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
echo "1:$i:4 " >> .machines
done
echo granularity:1 >> .machines
echo extrafine:1 >> .machines
...
Benchmark and scaling test on Tetralith
Here, I will demonstrate some results for running WIEN2k on the Tetralith cluster. The calculations below are using WIEN2k 18.2 with ELPA corresponding to the WIEN2k/18.2-nsc1-intel-2018a-eb module (though use the latest recommended one). They are for regular self-consistent DFT PBE calculations, setting RKMAX=7.
Disordered Ga2O3 with 120 inequivalent atoms, 1 k-point (normalized 1 job on 4 cores, 50401 s).
grep :RKM *.scf
:RKM : MATRIX SIZE 26231LOs:1032 RKM= 7.00 WEIGHT= 1.00 PGR:
Disordered NaCl with 64 inequivalent atoms, 1 k-point (normalized 1 job on 1 core, 2013 s).
grep :RKM *.scf
:RKM : MATRIX SIZE 5048LOs: 416 RKM= 7.00 WEIGHT= 1.00 PGR:
FeOCl with 12 inequivalent atoms, 416 irreducible k-points (normalized 1 job k-point parallel on 8 cores, 12563 s).
grep :RKM *.scf
:RKM : MATRIX SIZE 3049LOs: 184 RKM= 7.00 WEIGHT= 4.00 PGR:
The first two cases are for larger systems where 1 k-point might be suitable for the self-consistent calculations, while the third case is for a smaller system with more k-points.
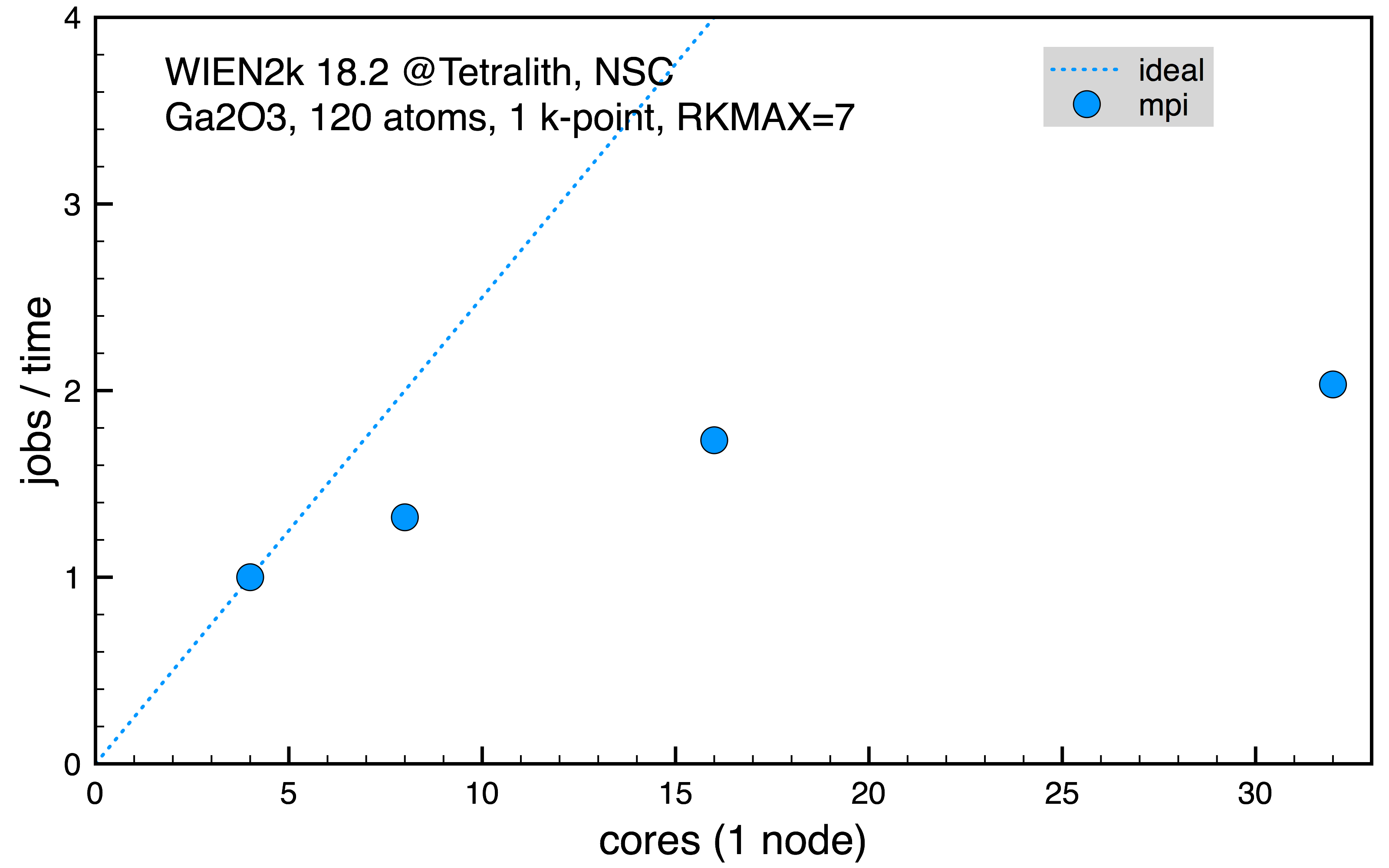
Fig. 1: Scaling on Tetralith Ga2O3 supercell with 120 atoms for 1 k-point.
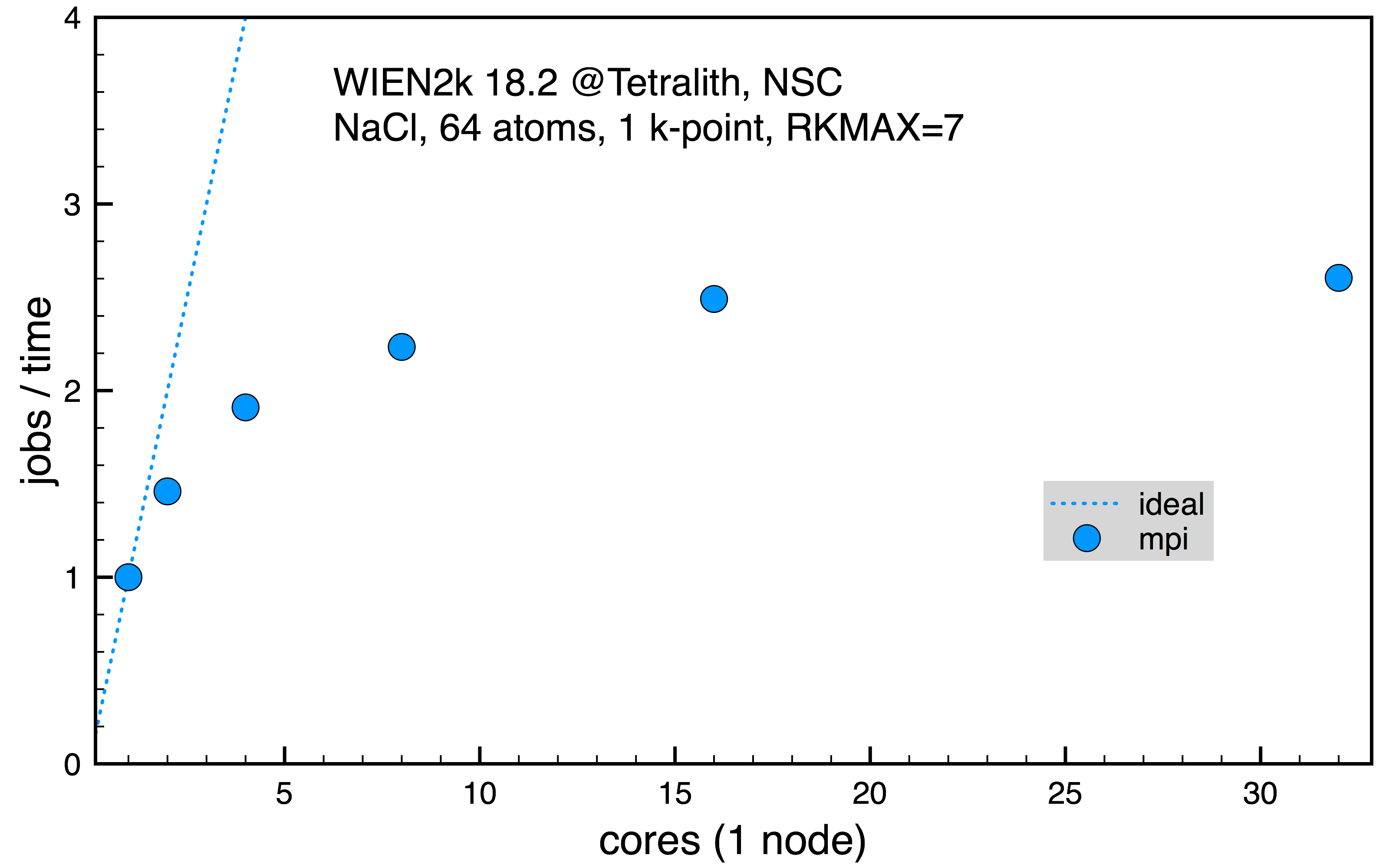
Fig. 2: Scaling on Tetralith for NaCl supercell with 64 atoms for 1 k-point.
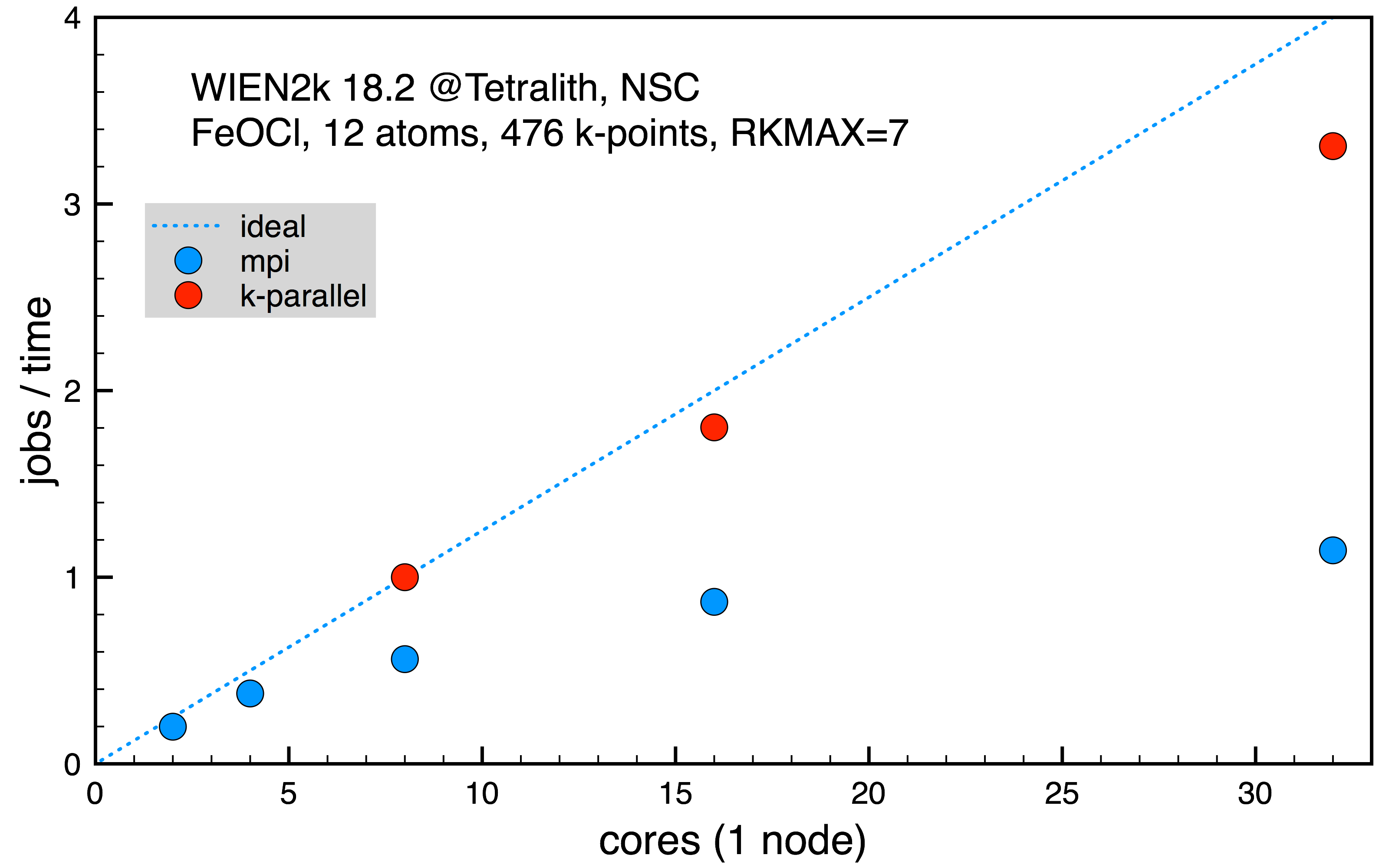
Fig. 3: Scaling on Tetralith for FeOCl supercell with 12 atoms for 416 k-points.
From the scaling in the first two cases, Fig. 1 and 2, it’s clear that increasing the number of cores in use for the mpi parallelism doesn’t increase the performance much. However, it might still be needed to allocate a full Tetralith node (32 cores) in order to have access to memory needed for the calculation. In such case, it’s still more efficient to run on 32 cores since it gives best performance, even if the scaling isn’t so good. In the last Fig. 3, the mpi parallelism is contrasted with the full k-point parallel scheme, effectively running several serial jobs. Here, it’s clear that it’s much more efficient to use k-point parallelism if possible.
 User Area
User Area

