Known limitations of MX software available in PReSTO
Here we list issues and features of MX software available in the PReSTO installation at NSC Tetralith and LUNARC Cosmos being relevant to be aware of as a user of these installations.
LUNARC Cosmos data processing
BioMAX data from Swedish academics will be automatically transfer to LUNARC Cosmos and available in /projects/maxiv/visitors/biomax/proposalNr however this directory is only available from the Cosmos login node. This is intentional and therefore data transfer from /projects/maxiv/visitors/biomax/proposalNr to home directory must be done prior to data processing using Cosmos compute nodes.
CryoSPARC jobs terminates due to GPU Usage Efficiency Policy at NSC Berzelius
Some CryoSPARC jobtypes such as 3D Classification does not meet the current GPU Usage Efficiency Policy decided at NSC Berzelius. Right now this is frustrating because NSC Berzelius performs extremely well during 3D refinement and other more compute intensive parts of CryoSPARC. PReSTO members from Swedish Cryo-EM community could approach the CryoSPARC developers asking them to deliver more efficient and parallel code during 3D Classification to avoid frustrating job terminations during 3D Classification. Since CryoSPARC is proprietary code, we cannot bluntly make these code updates without permission from the vendor. A potential workaround is using NVIDIA Multi-Instance GPU (MIG) that allows a single GPU to be partitioned into multiple smaller GPU instances - read more
Adding –reservation=safe to your SLURM command during 3D Classification enables you to avoid job termination. The safe reservation was recently introduced by NSC - more info.
License not available for CCDC/CSD in 2025
Members of the national CSD license in 2025 are SU, KTH, LU, UU, CTH, SLU and RICE. We hope that KI and LiU can join the national Swedish CSD license from 2026.
Useful info regarding CSD
Cambride Structural Database (CSD) is used by grade and grade2 from the BUSTER package to make ligand libraries for refinement. The CSD license system make compute nodes non eglible for CSD software packages so use login node when using grade/grade2 or CSD softwares such as Mogul/Mercury/GOLD etc.
The CSD license needs to be refreshed from time-to-time so if perhaps grade terminates with ModuleNotFoundError: No module named ‘ccdc’ please open a terminal window and perform ccdc_activator -a –activate -k XXXXXX-YYYYYY-ZZZZZZ-111111-222222-333333 and you are good to go for some time.
NanoPeakCell considered obsolete
NanoPeakCell version 0bce0c6 (git-commit number) is available in PReSTO but considered obsolete thanks to CrystFEL.
buster-report only runnable at Tetralith login node
While buster-report depend on CSD, buster-report only runs from the Tetralith login node. After a buster job has finished in a directory that we call buster3, a standar buster-report run would be:
module load BUSTER
buster-report -d buster3 -dreport buster3-report
Help and more options buster-report -h
CrystFEL 0.10 and HDF5 errors from GUI-loaded file lists
When supplying the CrystFEL GUI with a file list (of HDF5 data files), a failure to read the file list can occur. It appears that the CrystFEL GUI expects an event list (where each event/image within the HDF5 files is individually referenced). Such event lists can be generated with the CrystFEL (command line) utility “list_events” and should resolve the issue.
XDSAPP3 cannot handle spaces in PATH to .master file
XDSAPP3 is unable to handle spaces in PATH to .master file during “Load” step, however it can handle spaces in output directory.
How to process data from Diamond Light Source with XDSAPP3
XDSAPP3 expects image files named with data in their filename. It would be better if XDSAPP3 check into master.h5 to get the name of the image files because the information is there. Therefore, one cannot exchange crystal_100000X.h5 for crystal_1_data_00000X.h5 because then the info in crystal_1_master.h5 is incorrect and Durin crashes. Instead, one can do links so that XDSAPP3 internal parser get _data in the filename and Durin will also be happy.
In a terminal window enter directory with data and perform
for i in `seq 4`
do
ln -s "crystal_1_00000$i.h5" crystal_1_data_00000$i.h5
done
After soflinking the data can be processed normally!
DIALS run out of memory
Sometimes DIALS terminate with an out-of-memory statement like
Processing sweep SWEEP1 failed: dials.integrate subprocess failed with exitcode 1: see /native/1600/dials/DEFAULT/NATIVE/SWEEP1/integrate/12_dials.integrate_INTEGRATE.log for more details
Error: no Integrater implementations assigned for scaling
Please send the contents of xia2.txt, xia2-error.txt and xia2-debug.txt to:
xia2.support@gmail.com
slurmstepd: error: Detected 2 oom-kill event(s) in step 37678.batch cgroup. Some of your processes may have been killed by the cgroup out-of-memory handler.
DIALS software authors explain the issue/BUG in detail, and the current workaround is to use half of the available cores at various compute nodes i.e.
- use 16 instead of 32 cores available at NSC Tetralith compute nodes
- use 20 instead of 40 cores available at BioMAX offline-fe1 cluster compute nodes
- use 24 instead of 48 cores available at BioMAX clu0-fe-1 online cluster compute nodes
- use 24 instead of 48 cores available at LUNARC Cosmos compute nodes
For instance when using BioMAX offline-fe1 cluster with native.script change two lines of code into:
multiprocessing.nproc=20
sbatch -N1 --exclusive --ntasks-per-node=20 -J DIALS -o "$outdir/dials.out" --wrap="$dials"
Use one compute node for Phenix software
Queued jobs can use a single compute node only due to limitations in the code that needs to be redesigned in order for phenix to run on several nodes. A user can allocate more than a single compute node, however only a single node will be used for computing and the other nodes will simply be a waste of compute time. Eventually the improper multi-node phenix job will crash, however sometimes the job will finish and waste significant amounts of compute time that might be unnoticed to newcomers running phenix. Jobs can be submitted to the compute nodes directly from the phenix GUI running at the login node, however keep in mind only using a single node with 47 cores at Cosmos or 31 cores at Tetralith and adapt your allocation times accordingly.
Phenix mr_rosetta at BioMAX offline cluster
When running Phenix mr_rosetta at BioMAX offline-fe1 cluster the Log output window is blank during place model, however job is indeed running as more easily seen when reaching rosetta rebuild stage.
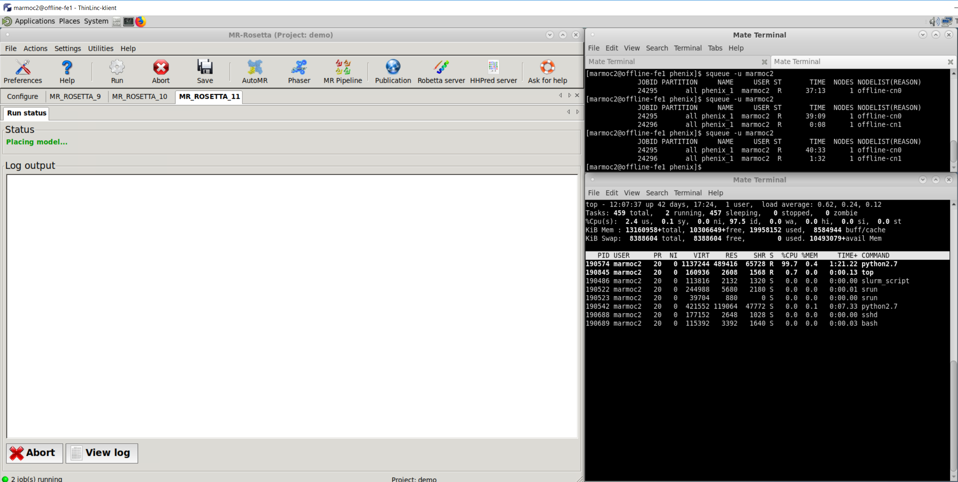 During Place model a blank Log output window is shown caused by logfile not created when GUI read it. This happen on BioMAX offline cluster running nfs filesystem in /home/DUOusername
During Place model a blank Log output window is shown caused by logfile not created when GUI read it. This happen on BioMAX offline cluster running nfs filesystem in /home/DUOusername
By using squeue -u DUOusername one can see that a job exists and by ssh offline-cn1 followed by top -u DUOusername that a process is running.
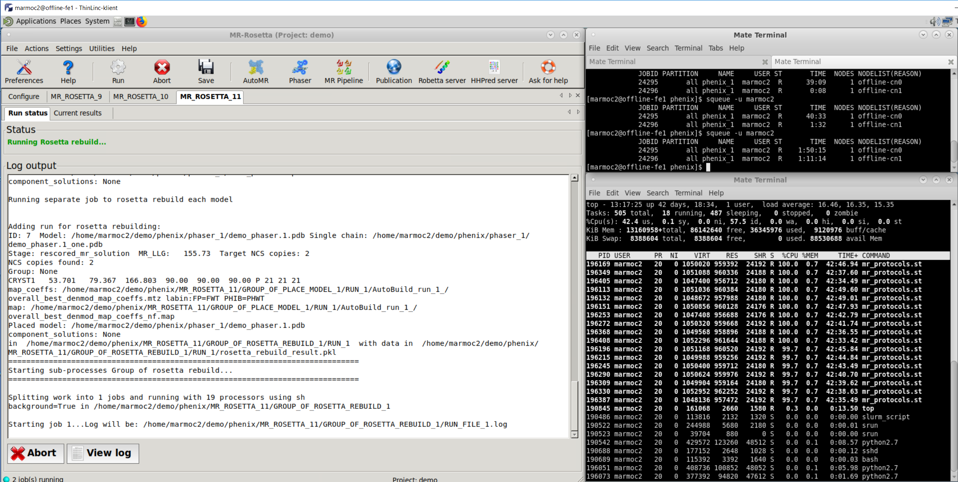 Here the job reached rosetta rebuild stage and many parallel processes are running in top terminal window
Here the job reached rosetta rebuild stage and many parallel processes are running in top terminal window
Use old path to rosetta as in Phenix 1.19.2-4158
Phenix developers recommend using Phenix 1.19.2-4158 with rosetta compared to using 1.20.1-4487. We can confirm this by successfully run a rosetta.refine test job using Phenix/1.19.2-4158-Rosetta-3.10-8-PReSTO that were indeed failing in Phenix/1.20.1-4487-Rosetta-3.10-5-PReSTO. The “path to Rosetta” setting in Phenix GUI should be: /software/presto/software/Phenix/1.19.2-4158-foss-2019b-Rosetta-3.10-8/rosetta-3.10
Phenix PHASER wizards login node running BUG
Phenix has two wizards for molecular replacement using PHASER software named Phaser-MR (simple one component interface) and Phaser-MR (full-featured). Neither of these two wizards send molecular replacement jobs to the compute nodes although Run-Submit queue job and Action-Queuing system-submit jobs… is available so this is a BUG in Phenix version 1.20.1-4487. The related MRage or Molecular replacement pipeline (advanced) has the same BUG when using Run-Submit queue job, however the Action-Queueing system-submit jobs… option is indeed sending molecular replcement jobs to the compute jobs as intended.
PyMOL/2.5.0-7-PReSTO contain SASpy ATSAS 3.1.0 plugin for PyMOL.
Later PyMOL versions depending on python3 does not contain the SASpy plugin.
 User Area
User Area

