

When I attended the supercomputing conference last year, I talked to many people in the booths and discovered that despite all the talk about petascale and exascale computing, VASP calculations are still a big part of what most supercomputing sites in the world are serving to their users. This seems to apply everywhere, not only in Sweden. But surprisingly, some described VASP as being “hugely problematic”, claiming that the parallel scalability was extremely bad – 64 cores maximum under any circumstances. In my experience, though, there is no problem running VASP on thousands of CPU cores, provided that you have a sufficiently large cell. My rule of thumb is that you need at least one atom per core, so if you have a supercell with a few thousand atoms, you can in fact run it with acceptable speed on current clusters by running it in a massive parallel fashion. It is true that a typical electronic structure calculation is not going to be that big, but I believe there are certain use cases for cells that big, for example when studying very low dopant concentrations, or simulations of nanostructures.
To show what would be possible for our users, provided they had a sufficiently large allocation of core hours, I decided to test the limits by setting up a new benchmark calculation: a 5900-atom supercell of Si-doped GaAs. It has dopants in random positions, and a big void in the middle, so there is no symmetry in this structure. In total, there is 5898 atoms, 23564 electrons, and about 14700 bands (depending on the number of cores employed).
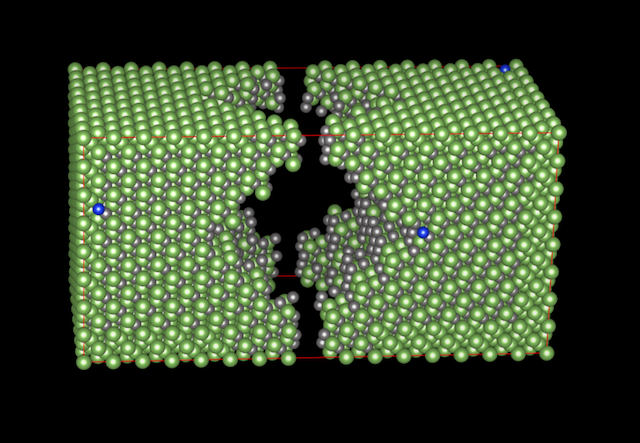
The questions at issue are: would you be able to run this cell with VASP given a big enough allocation on a Sweden HPC resource like Triolith or Lindgren; and from technical point of view, can you even run VASP on that many cores in the first place? The answer is “yes” to both questions, as is evident from the graph below. Employing 256 nodes and 3072 cores of Triolith gives us a speed of roughly 30 SCF iterations per hour, and with Lindgren, the number is 20 SCF steps/hour using 384 nodes and 9216 cores (i.e. 25% of the full machine).
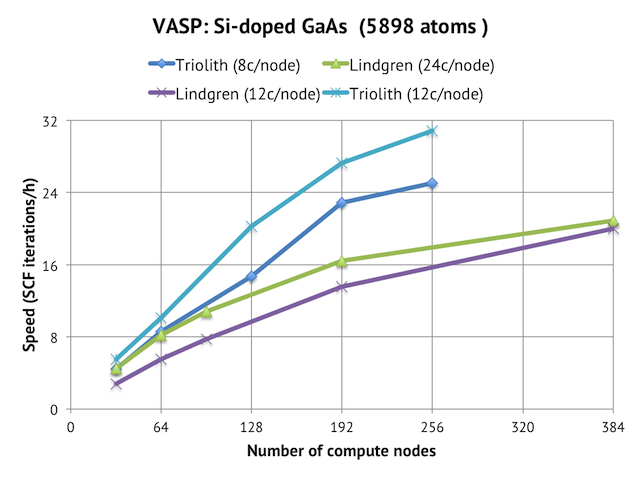
I would like to emphasize that no special magic is required to get a parallel scaling like that. It is the standard VASP source code compiled with the Intel toolchain, as described in the guides I have published. There were no special runtime settings other than NPAR=number of nodes and switching to RMM-DIIS exclusively (IALGO=48).
To illuminate in more detail, what is going on below the surface, we can look at the time profile of different subroutines. A regular VASP calculation will spend most of its time doing electronic minimization (subroutine RMM-DIIS or equivalent), but that is not what we find here. A breakdown from the 384-node run on Lindgren shows:
ORTCH (orthogonalization of wavefunctions) 40%
EDIAG (subspace diagonalization) 50%
RMM-DIIS (electronic minimization) 10%
Most of the time is spent orthogonalizing and deconstructing linear combinations of Kohn-Sham orbitals! At first, I suspected that this was a parallel scaling issue with SCALAPACK, similar to what I found for smaller systems, but the time profile is almost the same on 32 nodes, so evidently, those are real, new bottlenecks that come into play. The reason is, I believe, that ORTCH and EDIAG formally scales as N3, and that finally, they seem to have overtaken the other terms scaling as N2 and N2 log N. During the DFT world record (?) featuring 107292 atoms on the K computer, Hasegawa et al observed the same effect, with the conjugate-gradient minimizing part only being 1% of the computational cost.
To me, this proves that the computationally intensive parts of VASP are very well parallelized and that there are no serial parts that overtake the computations as the calculation is scaled up. The trick is rather that you need to scale up your systems as well, should you want to run big.
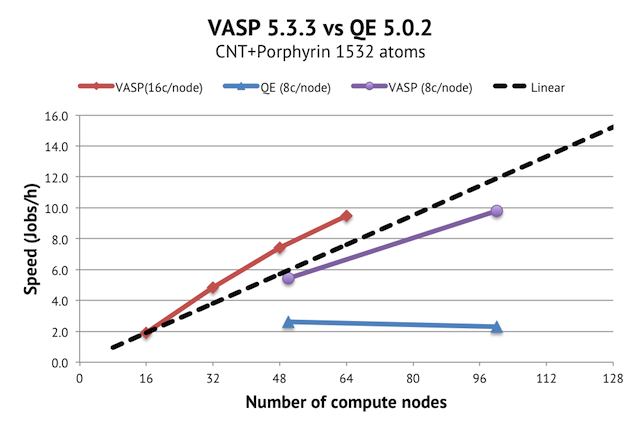
{kind=link}