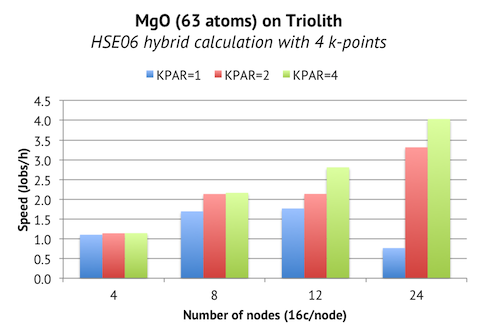
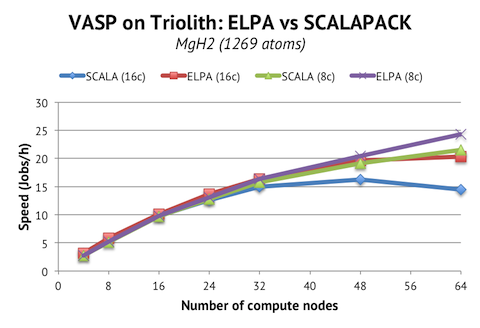
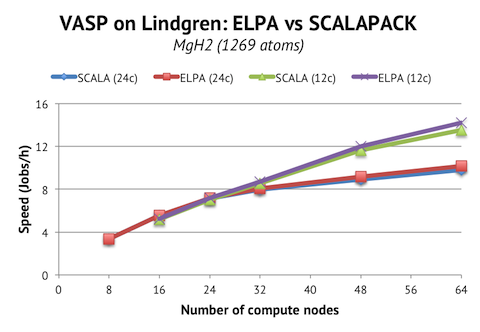
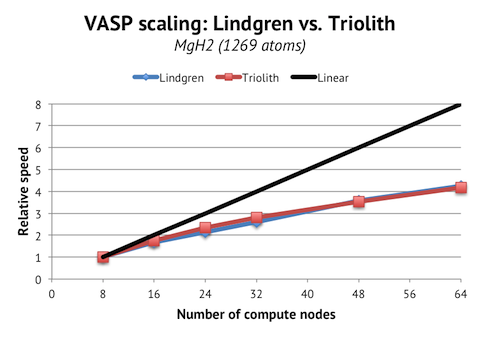
These instructions are for the 5.3.3 version, but I expect the instructions to be applicable to the minor versions preceding and following 5.3.3.
First, download the prerequisite source tarballs from the VASP home page:
http://www.vasp.at/
You need both the regular VASP source code, and the supporting “vasp 5” library:
vasp.5.3.3.tar.gz
vasp.5.lib.tar.gz
I suggest to make a new directory called e.g. vasp.5.3.3, where you download and expand them. You would type commands approximately like this:
mkdir 5.3.3
cd 5.3.3
(download)
tar zxvf vasp.5.3.3.tar.gz
tar zxvf vasp.5.lib.tar.gz
Currently, you want to load these modules:
intel/12.1.4
impi/4.0.3.008
mkl/10.3.10.319
Which you can get bundled in the following module:
module load build-environment/nsc-recommended
VASP 5 lib
Compiling the VASP 5 library is straightforward. It contains some timing and IO routines, necessary for VASP, and LINPACK. My heavy edited makefile looks like this:
.SUFFIXES: .inc .f .F
#-----------------------------------------------------------------------
# Makefile for VASP 5 library on Triolith
#-----------------------------------------------------------------------
# C-preprocessor
CPP = gcc -E -P -C -DLONGCHAR $*.F >$*.f
FC= ifort
CFLAGS = -O
FFLAGS = -Os -FI
FREE = -FR
DOBJ = preclib.o timing_.o derrf_.o dclock_.o diolib.o dlexlib.o drdatab.o
#-----------------------------------------------------------------------
# general rules
#-----------------------------------------------------------------------
libdmy.a: $(DOBJ) linpack_double.o
-rm libdmy.a
ar vq libdmy.a $(DOBJ)
linpack_double.o: linpack_double.f
$(FC) $(FFLAGS) $(NOFREE) -c linpack_double.f
.c.o:
$(CC) $(CFLAGS) -c $*.c
.F.o:
$(CPP)
$(FC) $(FFLAGS) $(FREE) $(INCS) -c $*.f
.F.f:
$(CPP)
.f.o:
$(FC) $(FFLAGS) $(FREE) $(INCS) -c $*.f
Note the addition of the “-DLONGCHAR” flag on the CPP line. It activates the longer input format for INCAR files, e.g. you can have MAGMOM lines with more than 256 characters. Now compile the library with the “make” command and check that you have the “libdmy.a” output file. Leave the file here, as the main VASP makefile will include it directly from here.
VASP 5 binary
Preparations
I only show how to build the parallel version with MPI and SCALAPACK here, as that is what you should run on Triolith. Navigate to the “vasp.5.3” library where the main source code is:
cd ..
cd vasp.5.3
Before we start, we want to think about how to find the external libraries that we need. These are:
- BLAS/LAPACK (for basic linear algebra)
- FFT library (for fast Fourier transform from reciprocal to real space)
- MPI (for parallel communication)
- SCALAPACK (for parallel linear algebra, e.g. orthogonalization of states)
For BLAS/LAPACK, we are going to use Intel’s Math Kernel Library (“MKL” henceforth). The easiest way to link to MKL at NSC is by adding the two following flags to the compiler command:
ifort -Nmkl -mkl=sequential ...
For fast Fourier transforms, we could use the common FFTW library with VASP, but MKL actually contains its own optimized FFTs together with an FFTW interface, so we can use these instead. Provided that we link with MKL, which we are already doing in order to get BLAS/LAPACK, we do not need to do anything more. The linker should pick up the FFTW subroutines automatically.
For MPI, we are going to use Intel’s MPI library. We have already loaded the “impi/4.0.3.008” module, so all we have to do is to add the “-Nmpi” flag to compiler command:
ifort -Nmpi ...
We don’t need to add explicit paths to any MPI libraries, or use the special “mpif90” compiler wrapper.
Editing the makefile
I suggest that you start from the Linux/Intel Fortran makefile:
cp makefile.linux_ifc_P4 makefile
It is important to realize that the makefile is split in two parts, and is intended to be used in an overriding fashion. If you don’t want to compile the serial version, you should enable the definitions of FC, CPP etc in the second half of the makefile to enable parallel compilation. These will then override the settings for the serial version.
Start by editing the Fortran compiler and its flags:
FC=ifort -I$(MKL_ROOT)/include/fftw
FFLAGS = -FR -lowercase -assume byterecl -Nmpi
We need to add “-Nmpi” to get proper linking with Intel MPI at NSC. Then, we change the optimization flags:
OFLAG=-O2 -ip -xavx
This is to be on the safe side, so that we get AVX optimizations. Include MKL with FFTW like this:
BLAS = -mkl=sequential
LAPACK =
We use the serial version of MKL, without any multithreading, as VASP runs MPI on all cores with great success. Set the NSC specific linking options for MKL and MPI:
LINK = -Nmkl -Nmpi
Uncomment the CPP section for the MPI parallel VASP:
CPP = $(CPP_) -DMPI -DHOST=\"LinuxIFC\" -DIFC \
-DCACHE_SIZE=4000 -DPGF90 -Davoidalloc -DNGZhalf \
-DMPI_BLOCK=8000 -Duse_collective -DscaLAPACK
-DRPROMU_DGEMV -DRACCMU_DGEMV
Change it to something like this:
CPP = $(CPP_) -DMPI -DHOST=\"TRIOLITH-BUILD01\" -DIFC \
-DCACHE_SIZE=4000 -DPGF90 -Davoidalloc -DNGZhalf \
-DMPI_BLOCK=262144 -Duse_collective -DscaLAPACK \
-DRPROMU_DGEMV -DRACCMU_DGEMV -DnoSTOPCAR
CACHE_SIZE is only relevant for Furth FFTs, which we do not use. The HOST variable is written out in the top of the OUTCAR file. It can be anything which helps you identify this compilation of VASP. The MPI_BLOCK variable needs to be set higher for best performance on Triolith. And finally, “noSTOPCAR” will disable the ability to stop a calculation by using the STOPCAR file. We do this to improve file I/O against the global file systems. (Otherwise, each VASP process will have to check this file for every SCF iteration.)
Finally, we enable SCALAPACK from MKL:
SCA= -lmkl_scalapack_lp64 -lmkl_blacs_intelmpi_lp64
And the parallelized version of the fast Fourier transforms with FFTW bindings:
FFT3D = fftmpiw.o fftmpi_map.o fftw3d.o fft3dlib.o
Note that we do not need to link to FFTW explicitly, since it is included in MKL. Finally, we uncomment the last library section:
LIB = -L../vasp.5.lib -ldmy \
../vasp.5.lib/linpack_double.o \
$(SCA) $(LAPACK) $(BLAS)
We have to do this to include the “$(SCA)” variable. The full makefile can be found here on Triolith:
/software/apps/vasp/5.3.3-18Dec12/build01/makefile
Compiling
VASP does not have a makefile that supports parallel compilation. So in order to compile we just do:
make
If you really want to speed it up, you can try something like:
make -j4; make -j4; make -j4; make -j4;
Run these commands repeatedly until all the compiler errors are cleared (or write a loop in the bash shell). Obviously, this approach only works if you have a makefile that you know works from the start. When finished, you should find a binary called “vasp”.
Running
When you compile according to these instructions, there is no need to set LD_LIBRARY_PATHs and such. Instead, the ifort compiler will hard-code all library paths by using the RPATH mechanism and write information into the binary file about which MPI version you used. This means that you can launch VASP directly like this in a job shell:
mpprun /path/to/vasp
Mpprun will automatically pick up the correct number of processor cores from the queue system and launch your vasp binary using Intel’s MPI launcher.