

VASP 5 introduced DFT hybrid functionals like PBE0. The Hartree-Fock calculations add a significant amount of computational time, however, and in addition, these algorithms require parallelization using NPAR=number of cores which is not as effective. In my experience, we are also haunted by SCF convergence problems, and you need to experiment with the other SCF algorithms. So what can we expect from Triolith here?
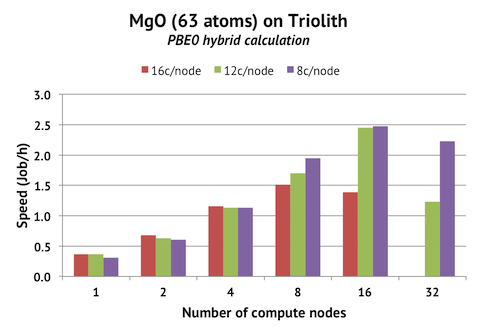
The chart above shows benchmark runs for a 63-atom MgO cell with Hartree-Fock turned on (corresponding to PBE0). ALGO=All is used, and NPAR=cores had to be set for each case separately. We find, not surprisingly, that we have good parallel scaling up to 4 compute nodes (equalling 1 atom per core). It is possible to crank up the speed by employing more compute nodes, but only by using 8-12 MPI ranks per node and idling half the cores. We have 192 bands in this calculation, so the maximum speed should be achieved with 16 nodes (16x12c/node = 192 ranks), which is also what we find (2.5 jobs/h).
These results should be compared with running the same job on Neolith, where an 16-node run (128 cores) reached 0.44 jobs/h, so Triolith is again a close to 6 times faster on a node-by-node basis.