

Please note that this is an old guide and not completely up-to-date. I am keeping it around mostly for reference. For up-to-date information, you should check one of the system-specific compile instructions and the VASP compiler status page.
Introduction
VASP is distributed as source code and everyone that works with VASP will have to rebuild the program from scratch sooner or later. Typical situations are: moving to a new supercomputer or cluster, activating certain nonstandard features of VASP, and performance optimization. This working paper collects some experiences of doing this on different computing platforms.
Prerequisites
To compile VASP, one needs a Unix-compatible system with:
- Standard Unix/Linux tools: make, tar, gunzip, cpp etc, i.e. you should be able to compile source tarballs from scratch. Most installations should support this. The exceptions might be “consumer” installations of some Linux distributions, and Mac OS X (one must install the Xcode development environment separately).
- A Fortran90-capable compiler (ifort, PGF, Pathscale, IBM XLF, gcc/gfortran have worked one time or another).
- The MPI parallel version requires an MPI installation, or at least the MPI development libraries and headers.
- BLAS and LAPACK libraries. An FFT-library is optional, because a decent one is included with VASP.
Getting and preparing the source code
The source of VASP comes in two files (“tarballs”) named something along the lines of: vasp.4.6.tar.gz, vasp.4.lib.gz. Usually one acquires these files from the official FTP server of the VASP Group.
[~/vasp]$ tar zxvf vasp.4.6.tar.gz
....
[~/vasp]$ tar zxvf vasp.4.lib.tar.gz
....
[~/vasp]$ ls
vasp.4.6 vasp.4.6.tar.gz vasp.4.lib vasp.4.lib.tar.gz
The folder vasp.4.6 contains the main source code of VASP, and vasp.4.lib contains a library with some subroutines necessary for compatibility. You can also find source files for lapack and linpack inside there.
Possible issue: Many VASP source files are meant to be preprocessed by cpp, they are named xxx.F, as opposed to the normal Fortran files that are named xxx.f. Obviously, this requires a case sensitive file system to work. This might be an issue on some Macs and old Unix systems (like OpenVMS). There is a straightforward workaround: edit the makefile to use .f90 instead of .f for preprocessed Fortran source files.
Building the VASP 4 library (libdmy)
Generally, the VASP 4 library builds without difficulties in most installations. There might be some complains about ranlib, but they are harmless. Create a new makefile by copying one of the template makefiles. They are called “makefile.something”. Use one that would seem to fit your computer system (makefile.linux for GNU/Linux etc).
[~/vasp]$ cd vasp.4.lib
[~/vasp/vasp.4.lib]$ cp makefile.linux makefile
[~/vasp/vasp.4.lib]$ nano makefile
[~/vasp/vasp.4.lib]$ make
Edit the lines containing FC and FFLAGS. These are for telling what Fortran compiler to use and which flags. Like this for the Intel compiler:
FC = ifort
...
FFLAGS = -O1
FREE = -free
or like this for gfortran:
FC = gfortran
...
FFLAGS = -O3
FREE = -ffree-form
In general, you would like to use a vendor supplied BLAS and LAPACK, so remove the lapack_double.o part from the makefile.
libdmy.a: $(DOBJ) lapack_double.o linpack_double.o
-rm libdmy.a
ar vq libdmy.a $(DOBJ)
When make has been run, you should be able to find a file called “libdmy.a” in this directory, if everything worked out ok. This library will later be used to build the vasp executable.
First build of VASP 4.6: basic version
The first aim is to manage to compile a version of VASP at all, i.e. no compilation errors, it should link correctly and run simple tests without crashing.
Finding the external libraries to use
VASP needs fast BLAS and LAPACK libraries. It is possible to download the source for these libraries from netlib.org and compile your own versions, but this is not the recommended way – they will be too slow. Instead one should use a library optimized for the particular hardware platform. The common choices are ACML (for AMD Opteron), MKL (for Intel cpu:s), GotoBLAS (multi-platform), ATLAS (multi-platform), ESSL (for IBM/PowerPC), Accelerate Framework (Mac OS X; based on ATLAS). You need to determine which one to use (see further notes below under Performance Optimization) and locate where it is installed in your computer system. If you are running at a supercomputer center, there is usually some kind of web documentation describing what is installed and where. If not, you should call support and/or look for libraries in standard places. These are: in the standard system directories for libraries, inside the compiler distribution, or in the site’s local software director.
Some examples below:
- Standard directories /usr/lib, /usr/local/lib, /lib
- Inside compiler distribution /opt/intel/mkl/10.0.3.020/lib/em64t, /sw/pgi/linux86-64/7.1/lib
- Local software /opt/gotoblas2, /sw/numlib/atlas, /software/acml4.0.1, /usr/local/lib
Once a library has been found, consult its documentation for how to use it. Sometimes several files need to be included for everything to work, and the linking order is very important. Especially Intel’s MKL is infamous in this matter. They even have a interactive web page for assisting you with linking MKL. It can also be a good idea to inspect the makefiles that come with VASP for clues on how to use the different BLAS and LAPACK libraries. E.g.: what have people used for ATLAS?
[~/vasp]$ cd vasp.4.6
[~/vasp/vasp.4.6]$ ls makefile.*
makefile.altrix makefile.hp makefile.linux_efc_itanium
makefile.linux_ifc_P4 makefile.rs6000 makefile.t3d
makefile.cray makefile.hpux_itanium makefile.linux_gfortran
...
[~/vasp/vasp.4.6]$ grep ATLAS makefile.linux_gfortran
#ATLASHOME= /usr/lib/blas/threaded-atlas
#BLAS= -L$(ATLASHOME) -lf77blas -latlas
#BLAS= $(ATLASHOME)/libf77blas.a $(ATLASHOME)/libatlas.a
This tells us that we will probably need to include two libraries for ATLAS to work: f77blas and atlas. (In fact, we also need to include ../vasp.4.lib/lapack_atlas.o)
The structure of the main VASP makefile
Most of the makefiles supplied with VASP have a special structure that is good to be familiar with when editing. In most cases, it is enough to copy an existing makefile and modify it, instead of writing your own from scratch. The general structure is:
- Comments with (sometimes) relevant info. Not all of the statements about certain libraries are up-to-date. Read for information and tips, but exercise caution.
- Settings for the serial version and libraries
- Settings for the parallel version (usually disabled by commenting)
- Main build part with specification of object files etc.
- Special rules (not always present). This section specify rules for compiling some of the source files with lower (or higher) optimization. Very aggressive optimization will break or degrade performance of some subroutines – these source files must be included in this section with different compiler flags.
The makefiles are laid out in a way that you will be able to use the same makefile for building both the serial and the parallel version. The idea is that first you make the appropriate changes in the serial section, build the serial executable, and then uncomment the lines in the parallel section (without changing the serial section), edit them, and build the parallel executable.
Basic settings
Again, start from the top and look for the lines containing FC and FFLAGS. This time, we will compile with no optimizations. The reason is that we want to exclude compiler optimizations as a source of error in case VASP does not work. It also makes compilation faster during the testing stage, which save time, because you may have to recompile everything several times before everything works. The following shows a relatively safe compilation mode for Intel Fortran compiler:
FC = ifort
FCL = $(FC)
...
FFLAGS = -free -fpe0 -fp-model strict -fpstkchk
...
OFLAG = -O0
The FFLAGS statements enforce strict floating point arithmetics and checks. The code will run slower, but at this stage we are only interested in getting it running at all. The OFLAG statement tells the compiler to not do any compiler optimizations. Other Fortran compilers have similar settings (inspect manual pages and documentation). If there are no flags for controlling floating point precision, at least specify the lowest optimization level. (Note: here it is very tempting to enable all sorts of compiler checks, such as array bounds checking. Doing this, however, may break VASP, because the source has not always been well-behaved in these aspects of sound programming).
Features – preprocessor flags
The next step is to review and edit the preprocessor flags. They are used to customize your VASP program for different platforms and job types. Look through the makefile for lines with “CPP = “ looking like this:
#-----------------------------------------------------------------------
# possible options for CPP:
# NGXhalf charge density reduced in X direction
# wNGXhalf gamma point only reduced in X direction
# avoidalloc avoid ALLOCATE if possible
# IFC work around some IFC bugs ugs
# CACHE_SIZE 1000 for PII,PIII, 5000 for Athlon, 8000 P4
# RPROMU_DGEMV use DGEMV instead of DGEMM in RPRO (usually faster)
# RACCMU_DGEMV use DGEMV instead of DGEMM in RACC (faster on P4)
#-----------------------------------------------------------------------
CPP = $(CPP_) -DHOST=\"MacXLF\" \
-Dkind8 -DNGXhalf -DCACHE_SIZE=2000 -Davoidalloc
There is often a commented (non-exhaustive) section above with some explanations of different choices. Consult the VASP manual for full descriptions. The syntax for each instruction is “-DKeyword”. Many of the options available here are advanced features and do not need to be used unless you are optimizing for maximum performance (see later section on optimization), but two of the settings are very important: NGXHALF and CACHE_SIZE. Enabling NGXhalf will reduce the memory requirements when running ordinary collinear calculations (i.e. no spin-orbit coupling). The option wNGXhalf reduces memory requirements even further, but only works for gamma point-only calculations, while NGXhalf works for arbitrary number of k-points. The standard way to compile VASP is to compile three different versions: one gamma-point only (with -DwNGXhalf and -DNGXhalf), one “half” version (with -DNGXhalf), and a “full” version for non-collinear calculations. Use the gamma-point only version for large supercells and molecules with only 1 k-point. It is roughly twice as fast and uses half the memory.
The second option that should be set is CACHE_SIZE. It sets the size of the cache memory used for calculating FFTs with Jürgen Furthmüller’s library. The size is given in number of 16-byte units (i.e. Fortran DOUBLE COMPLEX). A good starting guess for this number is to adapt the CACHE_SIZE to the size of the processor’s L1 data cache. (For optimal performance, one needs to make test runs). L1 cache information can be found by googling or visiting the processor manufacturers’ webpage (http://www.intel.com, http://www.amd.com, etc). Typical values range between CACHE_SIZE=1000 to 8000 (16KB to 128KB).
It is not necessary, but a nice gesture, to set the HOST flag to something else then what is already written in the makefile you use as template. VASP writes this information in the top of all OUTCAR files so that you can see which executable that was running. This applies especially if it contains someone else’s name. Put your own instead! Other useful information that can be put in the HOST flag is hostname and compiler used.
Compiling the serial version
It is now time to trying compiling using the makefile. Make sure that search paths for compilers and libraries are set up if it is necessary (e.g. PATH and LD_LIBRARY_PATH environment variables). Many compiler distributions include scripts to do this automatically.
[~/vasp/vasp.4.6]$ make
./preprocess <base.F | /usr/bin/cpp -P -C -traditional >base.f90 -DHOST=\"DarwinGCC\" -Dkind8 -DNGXhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
gfortran -ffree-form -ffree-line-length-none -O0 -c base.f90
./preprocess <mpi.F | /usr/bin/cpp -P -C -traditional >mpi.f90 -DHOST=\"DarwinGCC\" -Dkind8 -DNGXhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
gfortran -ffree-form -ffree-line-length-none -O0 -c mpi.f90
./preprocess <smart_allocate.F | /usr/bin/cpp -P -C -traditional >smart_allocate.f90 -DHOST=\"DarwinGCC\" -Dkind8 -DNGXhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
The building process will spew out lines looking like above (This one is from compiling VASP on with gfortan). The first line sends the .F files for preprocessing through cpp to generate a .f90 file. The second compiles the .f90 file with gfortran. It is repeated for all source files. If the compilation and linking finishes without errors, you should see last line that links all object files (*.o) together to form the executable:
gfortran -o vasp main.o base.o mpi.o smart_allocate.o xml.o constant.o jacobi.o main_mpi.o scala.o
asa.o lattice.o poscar.o ini.o setex.o radial.o pseudo.o mgrid.o mkpoints.o wave.o wave_mpi.o
symmetry.o symlib.o lattlib.o random.o nonl.o nonlr.o dfast.o choleski2.o mix.o charge.o xcgrad.o
xcspin.o potex1.o potex2.o metagga.o constrmag.o pot.o cl_shift.o force.o dos.o elf.o tet.o hamil.o
steep.o chain.o dyna.o relativistic.o LDApU.o sphpro.o paw.o us.o ebs.o wavpre.o wavpre_noio.o broyden.o
dynbr.o rmm-diis.o reader.o writer.o tutor.o xml_writer.o brent.o stufak.o fileio.o opergrid.o stepver.o
dipol.o xclib.o chgloc.o subrot.o optreal.o davidson.o edtest.o electron.o shm.o pardens.o
paircorrection.o optics.o constr_cell_relax.o stm.o finite_diff.o elpol.o setlocalpp.o aedens.o diolib.o
dlexlib.o drdatab.o fft3dfurth.o fft3dlib.o -L../vasp.4.lib -ldmy ../vasp.4.lib/linpack_double.o
-L/System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A -lblas -llapack
[~/vasp/vasp.4.6]$
You should now be able to find the executable file called “vasp” in the directory. It is a good idea to invent a scheme and rename this file so that you can keep different builds apart. Personally, I like to use the scheme [vasp]-[serial/mpi]-[build][XY]-[gamma/half/full]
[~/vasp/vasp.4.6]$ ls vasp
vasp
[~/vasp/vasp.4.6]$ mv vasp vasp-serial-build01-half
[~/vasp/vasp.4.6]$ cp makefile makefile.build01
This way, you can try several builds and have the binaries and the associated makefiles preserved.
Compiling the MPI parallel version
An MPI (Message Passing Interface) implementation needs to be installed on the computer system in order to compile (and run) the parallel version of VASP. Again, if you are using a supercomputer center, check if and where their MPI is installed. If different implementations are available, one is usually the recommended one. Nowadays, the popular implementations are “OpenMPI” (open source), “Intel MPI” (commercial), and “Scali MPI” (commercial). Special systems, like Cray’s may come with their own MPI library. On older systems, you might have some version of either “MPICH” or “LAM”. Beware that sometimes the installed MPI is compiled with Fortran77, which will creating linking errors related to underscores with VASP (which is compiled with Fortran90). Having an MPI compiled with Fortran90 makes life easier from the beginning.
To adapt the makefile for parallel compilation, look for the MPI section. It should be clearly marked in the makefile.
#=======================================================================
# MPI section, uncomment the following lines
#
We now need to uncomment and make changes to the lines containing FC, FCL, CPP and FFT3D. The idea in many of the makefiles provided with VASP is that these lines will overwrite the previous ones in the serial section. If there is no MPI section, make a copy of the serial makefile and edit the same lines. For OpenMPI, one should normally use the mpif90 Fortran compiler wrapper:
FC = mpif90
FCL = $(FC)
...
CPP = $(CPP_) -DMPI -DHOST=\"DarwinIntel\" \
-Dkind8 -DNGZhalf -DCACHE_SIZE=5000 -Davoidalloc
...
# FFT: fftmpi.o with fft3dlib of Juergen Furtmueller
FFT3D = fftmpi.o fftmpi_map.o fft3dlib.o
Note the addition of the “-DMPI” flag, and also that we should use “NGZhalf” instead of “NGXhalf”. In the FFT3D line, we changed from using the serial version of the FFT library to the MPI parallel one. There is usually no need to explicitly add the MPI libraries to the linking options, since we already use the mpif90 wrapper as the linking command, which will take care of this part automatically.
Just like before, we issue the “make” command, after cleaning up old object files. Example output from building parallel version on Mac OS X with gfortran 4.3.0 and openmpi 1.2.6:
[~/vasp/vasp.4.6]$ make clean
rm -f *.g *.f90 *.o *.L *.mod; touch *.F
[~/vasp/vasp.4.6]$ make
./preprocess <base.F | /usr/bin/cpp -P -C -traditional >base.f90 -DMPI -DHOST=\"DarwinGCC\" -Dkind8 -DNGZhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
mpif90 -ffree-form -ffree-line-length-none -O0 -c base.f90
./preprocess <mpi.F | /usr/bin/cpp -P -C -traditional >mpi.f90 -DMPI -DHOST=\"DarwinGCC\" -Dkind8 -DNGZhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
mpif90 -ffree-form -ffree-line-length-none -O0 -c mpi.f90
./preprocess <smart_allocate.F | /usr/bin/cpp -P -C -traditional >smart_allocate.f90 -DMPI -DHOST=\"DarwinGCC\" -Dkind8 -DNGZhalf -DCACHE_SIZE=2000 -DGfortran -Davoidalloc -DRPROMU_DGEMV
...
The executable file is called “vasp” as usual. We rename it, to keep it apart from the serial version.
[~/vasp/vasp.4.6]$ ls vasp
vasp
[~/vasp/vasp.4.6]$ mv vasp vasp-mpi-build02-half
To run VASP in parallel mode, you need to launch the executable using a special run command for MPI. For OpenMPI, it is called mpirun.
[~/vasp/vasptest/Tests/Si-cd]$ mpirun -np 4 ~/vasp/vasp.4.6/vasp-mpi-build02-half
The command above would run VASP on 4 cpu cores.
Testing the executables
Once you have compiled your first versions of VASP, it is time to test your compilations and verify the correctness of your executables. This is an important step. It is not good scientific practice to start running production calculations without having any idea if your “scientific instrument” is calibrated and is working correctly. Unfortunately, validating VASP is difficult because the source code (unlike many other ab initio programs) does not include either regression tests or functional tests. I consider this one of the weakest point of VASP. Possible ways to test your VASP installation are:
- Assuming that you trust your current production system, do regression testing with some previous calculations and compare output, especially things like total energies, forces and number of SCF iterations. Make sure that the tests cover features you would like to use, like different choices of ALGO, ISMEAR and LDA+U.
- Do functional testing by calculating some standard systems like Si(cd), Fe(bcc) etc, and compare with published values from experiments and other ab initio calculations. Make sure the calculations are converged well. If the values are close to what could be reasonably expected from DFT, chances are high that the program is working.
Save the test results you get, because you will need to repeat the tests and compare when doing performance optimization in the next chapter. The goal is to have the optimized version reproduce the same output as our safe compilation.
Note: I am currently making an automatic test suite for VASP (Link: here). It is yet far from comprehensive, but a passing mark should ensure, at least, basic operation of VASP.
Optimized build of VASP 4.6
Compiler optimizations and flags
It is tempting to spend a lot of time tinkering with different compiler optimization settings. This, however, is seldom time well spent, as the table below shows. Even the most basic optimization level gives almost all possible performance improvement (> 90%) from compiler optimizations. Later, we will learn to reason why. Many compilers have a flag for generating as fast code as possible, usually called “-fast” or similar. Otherwise, just try “-O” with the highest available level. You are unlikely to improve much upon these settings. Example makefile settings using Portland Group compiler, targeting AMD Opteron (rev E):
FC = pgf90
FCL = $(FC)
...
FFLAGS = -Mfree
...
OFLAG = -tp k8-64e -fast
Table 1. 10 SCF iterations of Fe13 icosahedral cluster (gamma-point only) using VASP executables compiled with gfortran 4.3.0 (2008-01-25) and different optimization settings. Running 2 MPI processes on 1.83 GHz Intel Core 2 Duo:
Optimization level |
Time |
Speed |
-O0 (no optimization) |
151 s |
1 |
-O1 |
107 s |
1.41 |
-O2 |
106 s |
1.42 |
-Os (-O2 but size restrictive) |
106 s |
1.42 |
-O3 |
105 s |
1.44 |
-O3 -ffast_math (unsafe) |
104 s |
1.45 |
In some cases, higher optimization will actually reduce performance or lead to numerical instability, like the example below:
Table 2. Same calculation as in Table 1. Intel Fortran Compiler 10 running with 2 MPI processes on 2.33 GHz Intel Xeon E5345 processor.
Optimization level |
Time |
Speed |
-O0 |
102 s |
1 |
-O1 |
33 s |
3.09 |
-O2 |
unstable |
|
-O1 / -O2 (some files) |
38 s |
2.68 |
-O3 |
unstable |
|
It is possible to circumvent this problem by putting special rules in the end of the makefile. They will enforce lower (or higher) optimization when compiling certain source files, to avoid breaking the sensitive source files. Many of the makefiles supplied with VASP have special rules of this kind. Look in the end of a makefile for the special rules section:
# special rules
# these special rules are cumulative (that is once failed in one
# compiler version, stays in the list forever)
#-----------------------------------------------------------------------
fft3dlib.o : fft3dlib.F
$(CPP)
$(F77) -FR -lowercase -O1 -c $*$(SUFFIX)
fft3dfurth.o : fft3dfurth.F
$(CPP)
$(F77) -FR -lowercase -O1 -c $*$(SUFFIX)
...
Here, “ff3dlib.F” and “fft3dfurth.F” and some other files are compiled with lower optimization (-O1), since -O2 and -O3 give worse performance, just like above. Again, since no regression tests are provided with VASP, pinpointing and diagnosing these kinds of compiler errors are very difficult. Sometimes, errors are obviously related to a single source file (e.g. corrupt XML output, try reducing optimization on xml.F and xml_writer.F), which you can then put in the special section for less aggressive optimization, but usually it is not that easy. You can use the rules in the existing makefile as clues before trying a brute force search (but don’t test one by one, use a divide-and-conquer approach).
Optimizing FFT “CACHE_SIZE”
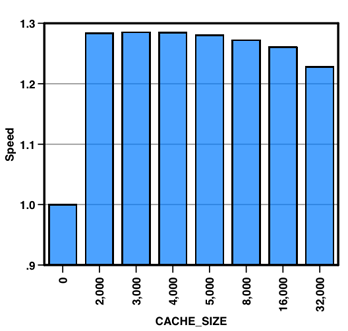
As mentioned previously, the preprocessor option “-DCACHE_SIZE” influences the data structures used for computing fast Fourier transforms. My experience is that the best value is usually the one that is close to the L1 data cache of the CPU used. The chart below shows VASP run times using different cache size values, tested on an Intel Xeon E5345 processor with 32 KB of L1 data cache per core. Based on that value, we would use the initial guess of CACHE_SIZE=2000 (2000*16 bytes ≈ 32 KB). This choice give almost optimal performance, but 3000 and 4000 are marginally faster. AMD Opterons have 64KB of L1 cache per core, and there I, and others, have found CACHE_SIZE=5000 to be the optimal choice. The most important thing is to not settle with CACHE_SIZE=0 just because you don’t know what value to use (as is suggested in the comments of the source code). At least test a couple of values like 0, 2000 and 5000.
Collecting data and profiling
“Premature optimization is the root of all evil” says the well-known proverb by Donald Knuth. Before we start to optimize our VASP program, we should know exactly in which subroutines the program spends its time, i.e what parts are in need of optimization. The best way to do this is to profile VASP running on your actual production system with a profiling program. Any decent supercomputing center will have such tools installed somewhere. If you are compiling a version for your laptop or workstation, you can use the GNU Profiler (“gprof”) or some vendor specific tool like Apple’s “Shark” profiler if you are on a Mac OS X system. As usual, it is good to profile both the optimized and unoptimized versions to get the complete picture.
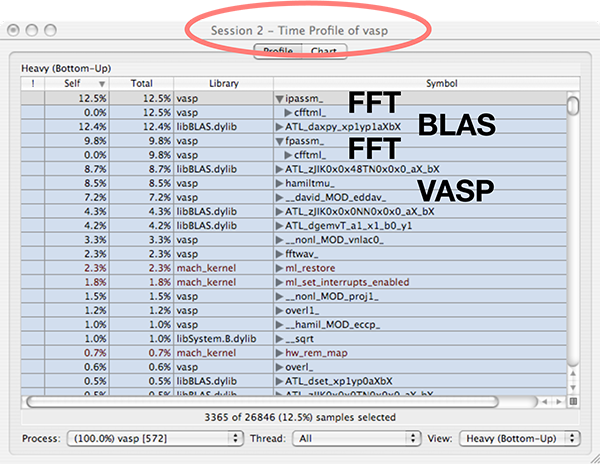
Below is shown the output from the Shark profiler when running an optimized version of VASP compiled with IBM XL Fortran (PowerPC architecture). The FFT library provided with VASP is used, together with the BLAS/LAPACK library provided with Mac OS X (based on ATLAS). Heavily used subroutines are listed in the top of the window. In the top, we mostly find calls to BLAS (libBLAS.dylib) and the FFT routines (ipassm, fpassm). They account for more than 80% of the runtime in this (very short) calculation. This is the explanation why we could not improve the speed of the code that much using compiler optimizations. The BLAS library is external to the VASP source code and is hopefully already optimized, and the FFT source code is not helped by aggressive compiler optimizations (-O1 or similar is enough). From this we draw the conclusion the performance of VASP is mainly determined by the choice of BLAS/LAPACK and FFT libraries. (This is of course also stated in the VASP manual). Which library that is most important depends on the exact system architecture and the size of the calculation that VASP is running. Usually, RMM-DIIS (which employs BLAS calls) will be the dominant subroutine in larger production calculations. There is a good discussion of scaling behavior in one of the original VASP papers (Kresse & Furthmüller, Phys. Rev. B 54, 1169).
BLAS & LAPACK
The easiest way to improve your VASP performance is to choose and test several BLAS/LAPACK installations. The general recommendations for best speed are:
MKL and ESSL are commercial products. The free, but not as fast, alternatives are GotoBLAS2 (no longer maintained?), ATLAS and ACML. You can usually make VASP work with all of these libraries, but I recommend staying away from ATLAS “developer” versions.
Below are some results from the Matter cluster at NSC (Xeon E5520 processors) running a medium sized 53-atom test job with VASP versions linked to different BLAS/LAPACK libraries (7-9 runs per data set):
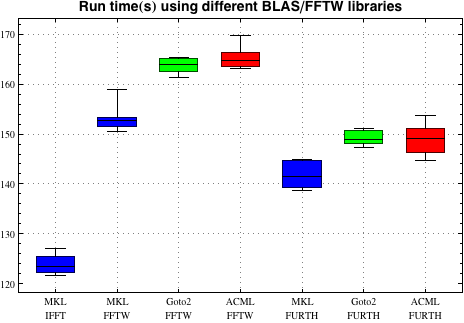
Here, the difference is quite dramatic. Using Intel MKL (including FFTs from MKL) produces a speed-up of about 30% compared to the slowest combination, with most of the speed gain coming from using the MKL FFTs. In isolation, ACML and GotoBLAS2 are both about 5% slower than MKL when compared using the same FFT library.
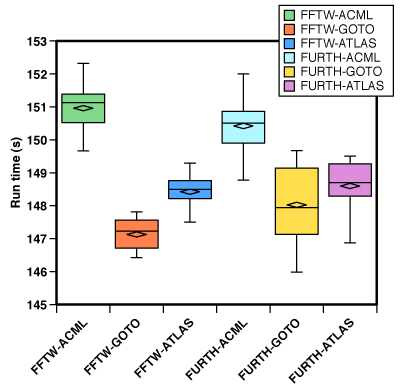
But sometimes, the results are not as conclusive. In the chart below, VASP was compiled on the (now retired) Isis cluster at UPPMAX with different numerical libraries. Isis had AMD Opteron rev. E processors. As above, 7 runs of each combination. The only statistically significant difference is that GOTO & ATLAS is faster than ACML, No statistically significant difference between FFTW and the built-in FFT library was found. Probably, Intel MKL should have been tested here as well, because recent tests on PDC’s Lindgren produced faster binaries with Intel MKL than the supplied Cray math library.
Caveat Do not make generalized conclusions from the numbers published here. The relative performance, and especially stability, of different math libraries often changes over time with new versions or new CPU:s. You really need to test on each new cluster. While big speed gains are sometimes possible, such as using Intel MKL on Intel processors, it must be emphasized that all BLAS/LAPACK libraries have been highly tuned by now, sometimes down to the point where one has to use statistical tests to able to distinguish test results. Still, 1-2% may translate into thousands of dollars in saved computer time for a large cluster installation.
FFT libraries
VASP uses fast Fourier transforms to switch between real space and reciprocal space. VASP has a built-in FFT library (“FURTH”) written by Jürgen Furthmüller which is very competitive with other libraries. The other choice is FFTW, or any other FFT library that can be called using the same interface (such as the FFT routines inside MKL).
The choice of FFT library is controlled by the FFT3D variable in the makefiles:
# FFT: fftmpi.o with fft3dlib of Juergen Furthmueller
FFT3D = fftmpi.o fftmpi_map.o fft3dlib.o
To switch to using FFTW, write instead
FFT3D = fftmpiw.o fftmpi_map.o fft3dlib.o /(path to FFTW library)/libfftw3.a
Note that you are using a different object file called “fftmpiw.o”. As usual, one needs to locate where FFTW is installed in the current computer system.
On machines with older Intel processors (P4/Core2 era), using FFTW gave a statistically significant increase in speed of about 1% on Intel systems, but on modern systems (Nehalem) FURTH is faster by ca 9%, when compared to FFTW 3.2.1. But as seen in the chart, using Intel’s FFT:s inside MKL is by far the best choice for speed.
No statistically significant difference was found on older Opteron systems.
For the Cray XE6 at PDC (with Opteron Magny-Cours chips), Intel MKL FFTs and FFTW are almost tied (FFTW ca 0.5% faster), and FURTH library is about 7% slower when compiled with Intel Fortran compiler.